Materials Science | ; Simulation, Learning
Predicting Ion Transport Kinetics at Complex Interfaces for Energy Storage
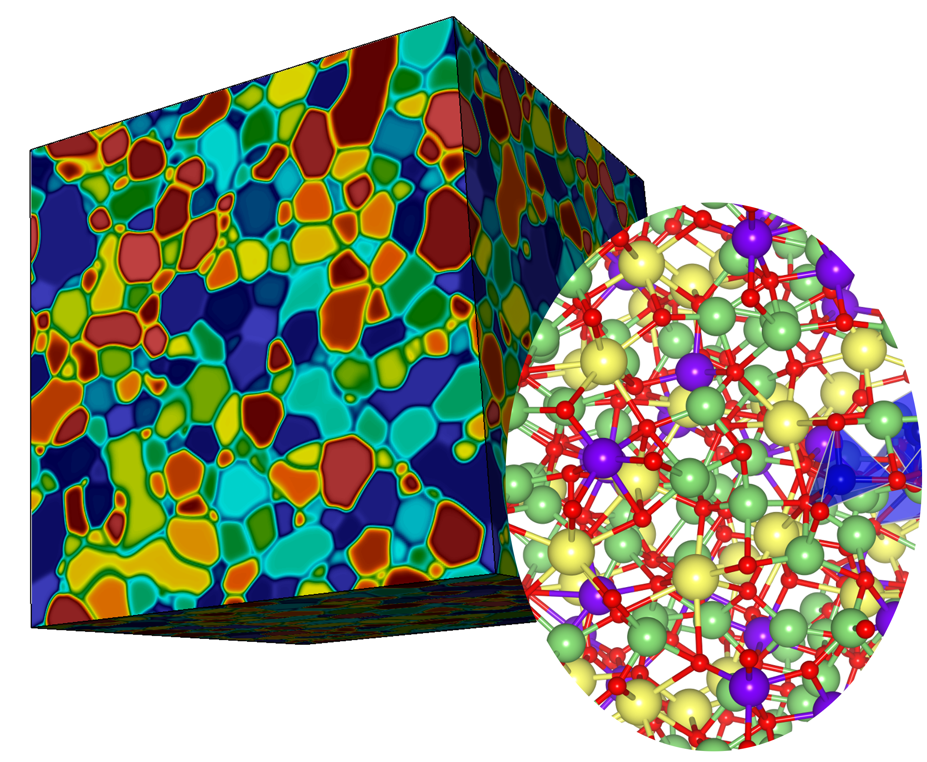
Many of the most promising high-capacity, solid-state energy storage systems—including next-generation batteries and hydrogen storage materials—rely on the rapid transport of ions through an electrolyte. This process, however, can be severely inhibited by complex and disordered interfaces in various devices. To shed light on this phenomenon, researchers from Lawrence Livermore National Laboratory are employing a multiscale modeling framework to predict ion transport kinetics at solid-solid interfaces.
Challenge
Because buried solid-state interfaces are notoriously difficult to probe experimentally—particularly during device operation—computational simulations play a unique and increasingly visible role. However, the idealized models often used for simulation studies neglect the chemical and structural disorder that real interfaces exhibit, in large part due to computational cost and complexity. As a result, interfacial ion transport is an exceptionally challenging problem to tackle using conventional approaches, instead requiring extensive multiscale simulations capable of spanning broad ranges of time and length scales.
Approach
Focusing on ceramic solid-state battery electrolytes and metal hydride hydrogen storage materials, this project integrates three sets of simulation capabilities to predict ion transport kinetics at interfaces. First, representative interface and disordered models are generated using large-scale ab initio molecular dynamics (AIMD) simulations, validated using computational spectroscopy. Second, these models are used as training sets for machine learning force fields able to span a much larger range of configuration space. Third, atomic-scale inputs are handed to a phase-field continuum model capable of simulating complex microstructures.
Results
In a study published in The Journal of Chemical Physics, the team employed machine learning to investigate Li7La3Zr2O12 (LLZO), a promising solid-state electrolyte material. The researchers developed and validated a machine learning potential for simulating crystalline, disordered, and amorphous LLZO systems across a wide range of conditions. Based on a neural network algorithm and trained using ab initio data, the machine learning potential was able to predict accurate structural and vibrational characteristics, elastic properties, and Li diffusivity of LLZO comparable to AIMD simulations. The team’s study demonstrated how this approach can enable simulations of transitions between well-defined and disordered structures with quantum-level accuracy at speeds thousands of times faster than traditional ab initio methods.
Impact
This project is harnessing supercomputing in combination with advanced machine learning techniques to simulate ion transport kinetics in novel energy storage systems with unprecedented accuracy. With a better understanding of how interfaces impact ion transport, researchers can develop rational engineering strategies for improving performance of advanced battery materials for grid and vehicular energy storage applications.