Biological Sciences | Simulation, Data, Learning
COMPBIO2: Combining Deep Learning with Physics-Based Affinity Estimation 2
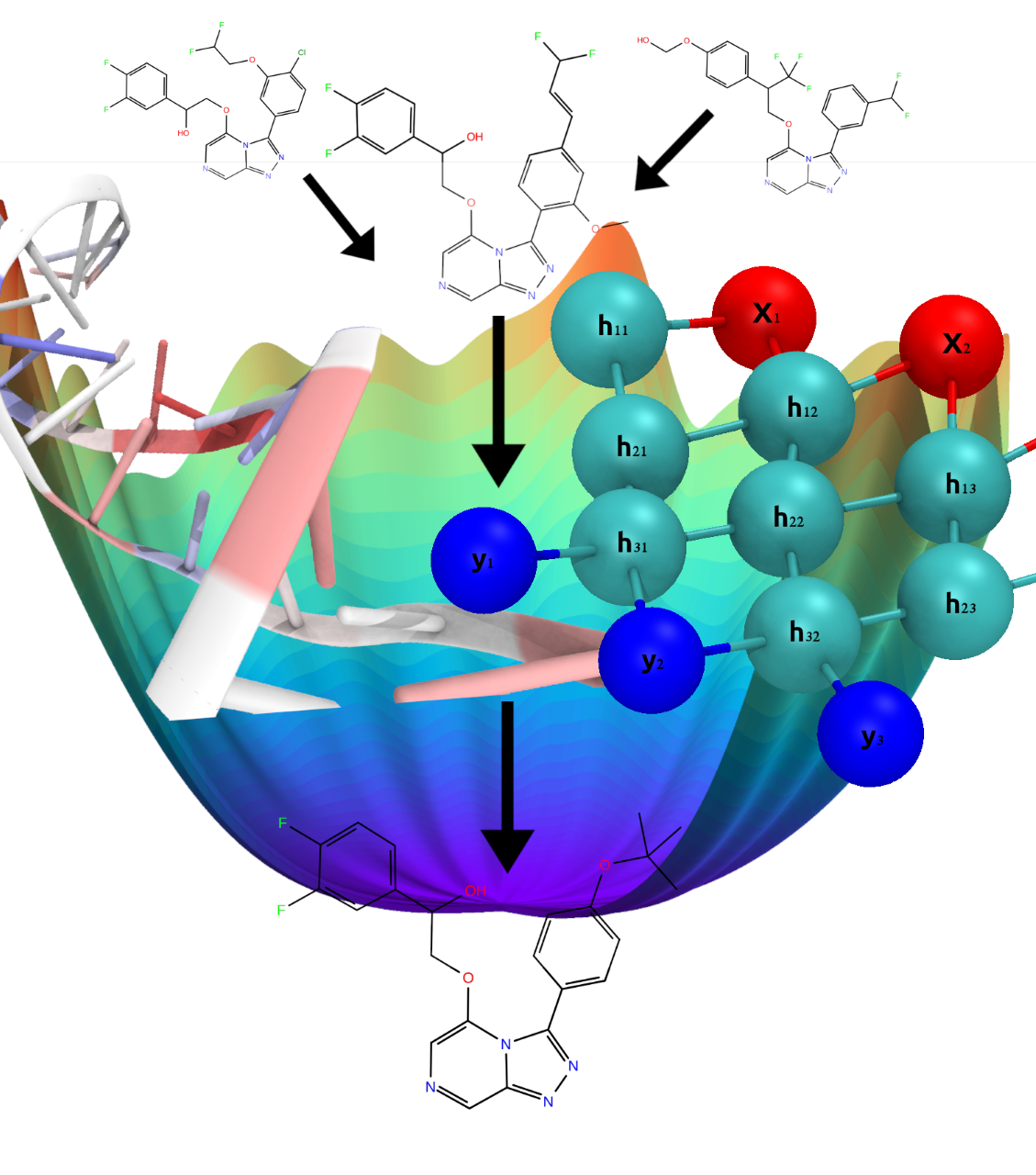
Machine learning and physics-based methods combined can dramatically accelerate the process of drug discovery. With this INCITE project, researchers are developing computational methods to overcome severe bottlenecks in the pharmaceutical industry’s current process and speed the search for drug candidates for SARS-CoV-2 and other viruses.
Challenge
Drug discovery requires exploring a vast chemical space of potential ligands to test binding potency with target proteins. In silico (computer simulation) physics-based methods play a key role in computational drug discovery but there is a very high computational cost when using relative binding free energy (RBFE) methods based on classical molecular dynamics (MD) which can lead to insufficient sampling of the chemical space (i.e., looking at too small of a subset of possibilities). The other, less computationally expensive method of finding drug binding potency is through deep learning, which requires a huge amount of training data. This project combines both to overcome limitations of each method.
Approach
This project combines deep learning with physics-based methods and employs a range of ensemble simulation-based methods. The most accurate among them is called Thermodynamic Integration with Enhanced Sampling (TIES) which performs an ensemble of MD simulations at each alchemical intermediate state and then integrates the ensemble averaged energy derivative to obtain RBFEs. The pipeline using AI and simulation to look for drug candidates for COVID-19 is known as IMPECCABLE (Integrated Modeling PipelinE for Covid Cure by Assessing Better Leads). ALCF staff worked with the team to build NAMD code and helped implement the sampling enhancement method REST2 on NAMD CUDA.
Results
The team was able to simulate many more ligand transformations with greater accuracy, reproducibility, and precision than general simulation methodology. With TIES, the team replicated MD simulations in each free energy window five times with a different initial velocity allowing for statistically meaningful results and complete control of errors based on direct ensemble averages. They computed a broad selection of over 500 ligand transformations involving over 300 ligands binding to 14 different protein targets across a wide range of protein classes and found excellent correlation between experimental and prediction results. The systematic and extensive analysis of these transformations over such a large dataset is unprecedented.
Impact
The team has demonstrated a method to overcome the limitations of physics-based drug simulations, allowing more accurate and reproducible results that can substantially reduce the time and cost of bringing new drugs to market. The team’s method will also enable researchers to assess the resistance of COVID-19 protein variants and their impact on existing drugs and vaccines.
PUBLICATIONS
- “Large Scale Study of Ligand-Protein Relative Binding Free Energy Calculations: Actionable Predictions from Statistically Robust Protocols,” Journal of Chemical Theory and Computation (March 2022), ACS Publications.
- “Alchemical Free Energy Estimators and Molecular Dynamics Engines: Accuracy, Precision and Reproducibility,” Journal of Chemical Theory and Computation (May 2022), ACS Publications.
- “Ensemble Simulations and Experimental Free Energy Distributions: Evaluation and Characterization of Isoxazole Amides as SMYD3 Inhibitors,” Journal of Chemical Theory and Computation (May 2022), ACS Publications.